Retinitis Pigmentosa (RP) refers to a group of eye conditions that affect the retina, and are caused by a genetic predisposition. It is estimated to affect around two million people around the world, and can often run in families – although it is not unusual for only one person from a family to be affected. It is usually detected in childhood or adolescence, and can progress over the entire course of a person’s life, or may halt in progress at some point.
The retina is the light-sensitive, innermost layer of the eye that is responsible for processing images received by the eye and transferring them to the brain as nerve impulses in order to create visual perception. It is rich in photoreceptor cells which are of two main types – rods and cones. Rods are involved in dim light and monochromatic vision, while cones are involved in bright light and coloured vision. The genetic mutations that cause RP prevent these cells from functioning properly.
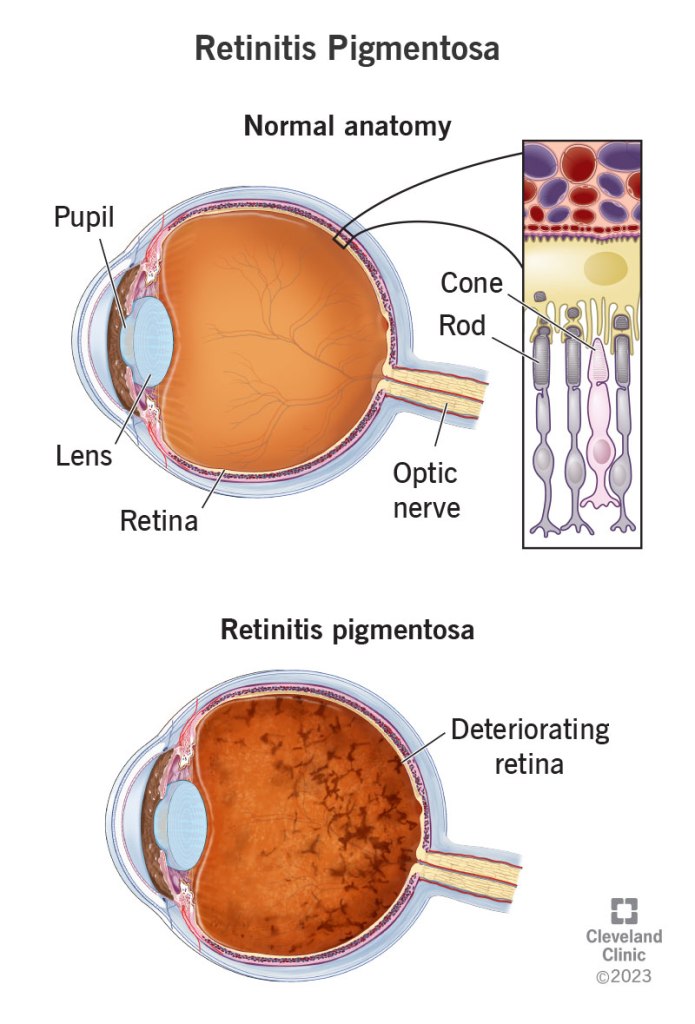
Illustration from Cleveland Clinic
Symptoms
In most cases, the genetic mutations that cause RP are such that it is initially (or only) the rod photoreceptor cells that get affected. Hence, one of the first symptoms that might be noticed is decline in or loss of night vision. Cones are concentrated towards the center of the eye, whereas rods are found more to the sides. Therefore, another symptom that might be noticeable is loss of peripheral (side) vision. This is referred to as tunnel vision. That is, vision from the corner of the eye declines. Usually, this happens concurrently in both eyes.

Image from MyVision.org
If cone photoreceptor cells get affected over time, this may lead to symptoms such as sensitivity to light and decline in or loss of colour vision, as well as potential decline in or loss of central vision.
Complications
RP is generally a progressive disease, which means that people with RP will experience a progressive decline in vision over time. This is usually mostly observed in the form of reduced peripheral vision and possibly, in some cases, eventual blindness. The progressive nature of the condition could thus impact the quality of life of the person concerned, bringing about limitations to their scope for work and independence over time.
Additionally, individuals with RP may have a higher risk of developing early-onset cataract and macular oedema (retinal swelling).
Causes
As mentioned above, RP is a hereditary condition, and gets passed on from parent to child. Symptoms may not always manifest in every generation, but one who bears the mutation causing RP maybe a carrier for it for the next generation. This is particularly the case when the mutant gene is carried by the mother (X-linked inheritance), as the condition may only be mildly expressed phenotypically (physically) in about 1 in 5 affected females. Male offspring, however, may be affected.
In rare cases, RP may also be caused by some medicines, eye infection or injuries, or may be linked to other genetic conditions such as Usher Syndrome, which causes both hearing and vision loss.
Diagnosis
There are several tests that might be used by an ophthalmologist to confirm an RP diagnosis. These include:
- visual field testing – to measure peripheral vision and keep tabs on the development of blind spots
- electroretinography (ERG) – to measure the retina’s response to light
- optical coherence tomography (OCT) – to determine how exactly RP is affecting the retina
- genetic testing – to determine the presence of RP-causing mutant genes
Ideally, diagnosis of RP should be followed by regular eye check-ups in order to determine the nature of progress of the condition in order to best manage it. Healthcare professionals recommend that family members of patients also get screened for RP as it is a hereditary condition.
Prevention
Being hereditary in nature, there appears to be no way of preventing the incidence of RP. If someone who intends to have children is known to have RP, they may choose to seek genetic counselling in order to get a better idea of the risk of passing it on to the next generation.
A few measures that can be taken to rein back worsening of the condition include protecting eyes from sunlight UV rays (such as using suitable sunglasses), and avoiding overconsumption of vitamin E (limit to 30 IU per day). Contrary to previous belief, high consumption of vitamin A does not seem to significantly benefit the condition in any way.
Treatment
At the moment, there is no known treatment for the alleviation of RP symptoms or to stop its progress as there are over 100 possible gene mutations that could give rise to the condition. However, extensive research is being carried out on RP cases and their families with the hope of developing some form of treatment – most likely gene therapy – to be able to assist patients with this condition in the future.
Patients with RP may receive guidance from vision specialists on tools and techniques that may be useful to help deal with and minimise the impact of progressive vision loss on their day-to-day life.
Cover illustration adapted from Freethink
